
Medical equipment lifecycle management
Medical equipment lifecycle management of re-usable medical equipment can be effectively summarised in the following diagram.
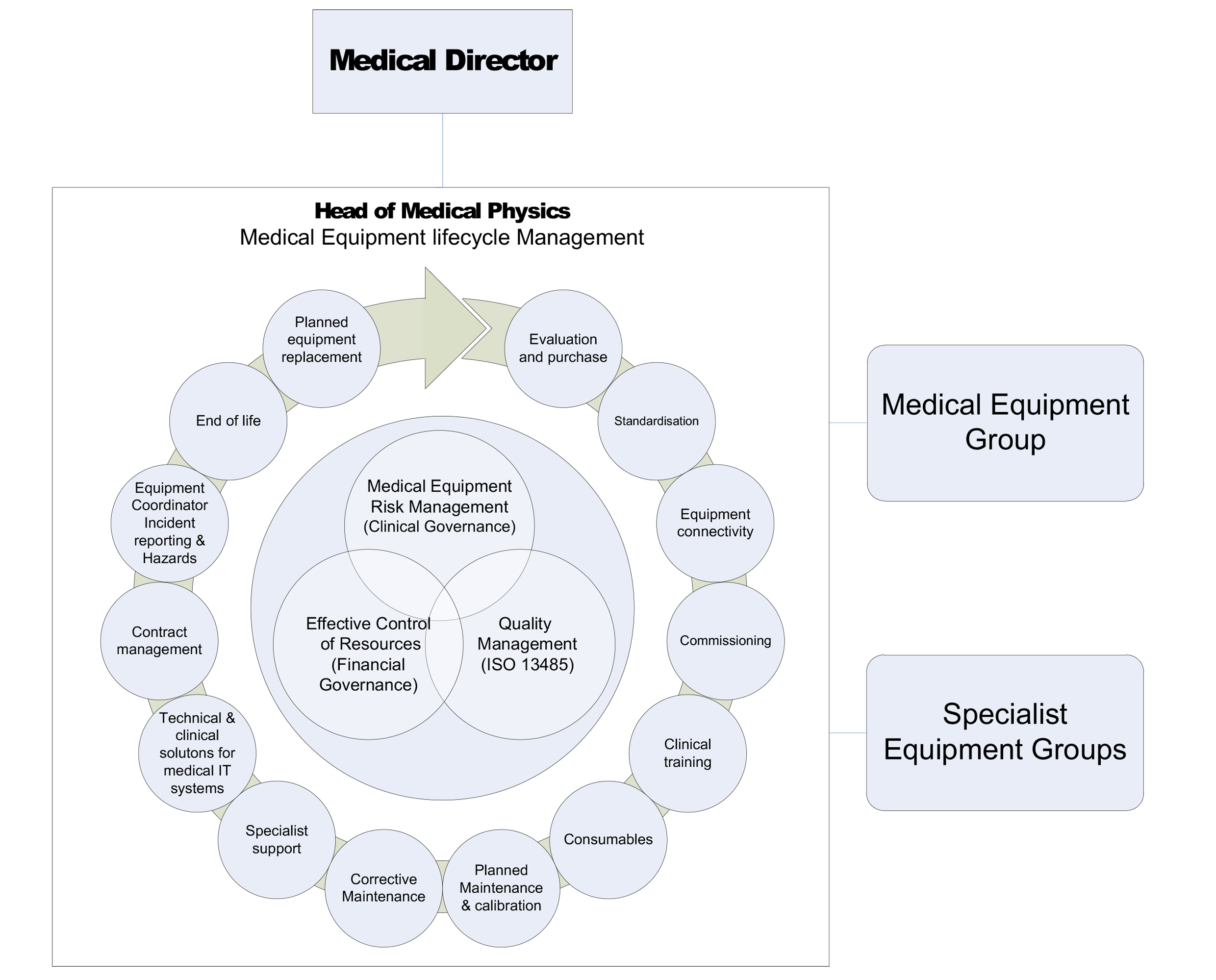
Selection and procurement
The process for the selection and procurement of reusable medical equipment is defined in the Board’s policy.
CS-MPH-POL-10, Medical Equipment Funding and Acquisition Policy
The process is overseen by Materials Management with appropriate expert advice and direction provided on an ongoing basis. It is designed to eliminate rogue purchases and ensure appropriate stakeholder involvement. The process ensures Infection Control input to all medical Equipment purchases. Tenders for high value procurements include assessment of environmental and sustainability credentials of equipment suppliers and the medical equipment under consideration.
Standardisation and connectivity
The procurement process ensures that consideration is given to equipment standardisation and connectivity. The main driving force for equipment standardisation is the management of risks associated with equipment usage (training, familiarity, reducing variation). It also minimises maintenance and consumable costs, reducing overall lifecycle spend. Standardisation also enables a high level of connectivity and integration between devices to be effectively implemented.
Commissioning
It is essential that all new equipment is checked against specification and receives safety checks before being placed into clinical use. A dedicated acceptance check procedure exists to define and standardise the commissioning process.
CS-MPH-PRO-12, Acceptance Check Procedure
The procedure describes:
- Delivery checks
- Pre-use checks
- Electrical Safety checks
- Storage during commissioning
- Record keeping
- Documentation management
Electronic inventory
The commissioning process requires important equipment management information relating to medical equipment to be stored on an electronic inventory. Good record keeping underpins the safe and effective management of medical devices. The Medical Physics Database is a centralised equipment management database designed to allow the Board to appropriately administer its medical devices. The database meets all the requirements and minimum datasets set out in guidance by the MHRA and the Scottish Government. Functions of the database include:
- Electronic inventory
- Planned preventative maintenance management
- Corrective maintenance management
- Customer request management
- Contract management
- Planned equipment replacement
- Engineer dashboards
- Supplier administration
- KPI and customer metric generation
- Finance – asset register verification
A second parallel inventory of capital medical equipment exists in the Finance Asset Register.
Documentation (Instructions for use)
Good clear instructions for use have a crucial role in the continued safe and effective use of medical equipment. The lack of adequate instructions for use is recognised by the MHRA as a key equipment safety consideration and a root cause of adverse events.
When a new equipment type is first introduced to a department, a copy of the instructions for use is provided by Medical Physics. In addition to this, a central library of all instructions for use documents is held and maintained by the Medical Physics department. All users have access to the documents on request. Additional copies can also be made available. The process for the distribution of instructions for use is included in the Medical Physics acceptance check procedure.
Training
Clinical user training
All healthcare professionals working with medical equipment have a professional duty to ensure their own skills and training remain up to date. Training is a key element in device safety. The quality of training in the appropriate use of medical devices is a recognised root cause in the occurrence adverse incidents. All professional groups within the Board with a responsibility for the safe use of medical devices must have their own medical equipment training policies. The policies must define the requirement and process for adequate record keeping.
Technical training
Members of the Medical Physics department directly responsible for the repair and maintenance of medical equipment must be trained, qualified and have the necessary levels of competency to meet the operational requirements of the department. A dedicated training procedure exists to ensure that this requirement is met at all times.
CS-MPH-PRO-10, Training Procedure (Medical Physics)
The procedure describes the process for:
- Competency assessment
- Identifying training needs
- Departmental requirements to meet service delivery
- New members of staff
- New equipment
- Producing a training plan
- Recording training on individual and departmental training matrixes
Maintenance and repair
Medical device maintenance and repair falls into two distinct categories, corrective maintenance and planned preventative maintenance. Corrective maintenance occurs when a device fails unexpectedly. The process for dealing with these failures ensures a swift response and resolution to any problems.
CS-MPH-PRO-4, Corrective Maintenance Procedure
Planned Preventative Maintenance (PPM) is managed routine maintenance, scheduled to take place at defined intervals and intended to prevent failures from occurring in the first place.
CS-MPH-PRO-8, PPM of Electromedical Equipment Procedure
Decontamination of medical equipment
Decontamination of medical equipment is covered thoroughly by the Board’s infection control policies. All staff have a responsibility to ensure equipment is decontaminated before it is received by Medical Physics. All requests for corrective maintenance should be accompanied by a combined request/decontamination form.
Specific guidance for Medical Physics staff is included in the departmental policy.
CS-MPH-PRO-17, Infection Control Procedure (Medical Physics)
Tracking
All maintenance operations are tracked by the call log system within the Medical Physics Database. All equipment undergoing repair is accompanied by a status label to indicate the equipment fault.
Storage (control of product)
Robust control of product is essential to the safe application of the maintenance process. It is common for equipment to be in varying states of repair when stored in Medical Physics labs. It is essential that entry to the labs is controlled to ensure equipment is not disturbed or returned to use before repair is complete.
Specialist support
The Medical Physics Department operates a single department structure across multiple clinical/technical specialities. This differs from the common model of operating multiple specialist departments (anaesthesia, cardiology, renal etc). The range of equipment covered is supported by Specialist Engineers in key specialities. These lead Engineers require more in-depth training and knowledge to address the specific requirements of this equipment and its clinical application.
Device manufacture and modification by Medical Physics
The in-house manufacture of medical devices is not included within the Medical Physics quality management system. Device manufacture will only be considered in exceptional circumstances when an equivalent device is not commercially available.
The MHRA Provides specific guidance on custom made devices in
In house manufactured devices are exempt from CE Marking as long as they are only used within the manufacturing organisation. However, a robust formal risk assessment will be required to ensure:
- Electrical safety
- The device is cleanable
- Appropriate consideration is given to what happens if it fails
- Appropriate ‘conditions of use’ documentation is produced
- Controls are in place to ensure the equipment is only used within Board premises
Planned equipment replacement
The information held on the Medical Physics database is used to drive the planned equipment replacement programme. The programme generates a 10-year outline financial projection which is used to inform medium to long term capital planning.
One of the core remits of the Medical Equipment Group (MEG) is to oversee all medical equipment purchases in the context of clinical strategies. The group has strong clinical representation across all directorates and clinical specialities. Annual replacement lists derived from the planned equipment replacement plan are verified by the group before going on to form part of the Board’s annual capital plan.
The replacement programme also allows additional individual factors to be considered outwith the planned element of replacement. These are laid out in the ‘medical equipment purchase process’ and include whether the item is:
- Worn out beyond economic repair
- Damaged beyond economic repair
- Unreliable (based on service history)
- Clinically or technically obsolete
- Spare parts are no longer available
- Superseded by a more cost effective or clinically effective device
- Unable to be cleaned or decontaminated effectively
End of life (decommissioning and disposal)
A process for the removal from service and disposal of medical equipment is essential element of medical equipment lifecycle management. Equipment is regularly removed from use for a verity of reasons including:
- Equipment replaced as part of the planned equipment replacement programme
- Damaged or worn out beyond economic repair
- Clinical or technical obsolescence
- Disposal due to contamination, e.g. Creutzfeldt-Jakob (CJD) etc.
- Changes in local policies for device use
- Absence of manufacturer/supplier support
- Non-availability of correct replacement parts
- Non-availability of specialist repair knowledge
A dedicated procedure exists for the decommissioning and disposal of medical equipment.
CS-MPH-PRO-3, RTM Procedure
The procedure ensures compliance with the WEEE directive. A separate procedure is in place for the disposal of media containing sensitive data.
CS-MPH-PRO-35, Disposal of Media Containing Sensitive Data
Governance
Hazard notifications
For the purposes of this policy, the terms Hazard Notification. Safety Notice and Medical Device Alert are interchangeable.
Hazard notifications can originate from the MHRA, HFS, equipment suppliers, equipment manufacturers or can be internally generated by Medical Physics. Distribution and tracking of hazard notifications is the responsibility of the Equipment Co-ordinator (Head of Medical Physics). The process for handling hazard notifications is defined in the following policy.
CS-MPH-POL-5, Hazard and Safety Action Notice Policy
Medical equipment incidents
All incidents or near misses relating to medical equipment must be processed in line with the Board’s standard incident reporting policy.
COR-CG-RM-POL-5, Incident Reporting Policy and Procedure
Incidents involving medical equipment must also be reported to Medical Physics immediately to ensure prompt investigation. Where possible, all material evidence relating to adverse events must be preserved, labelled and kept secure. This includes the medical device, consumables, packaging and any other means of batch identification. Equipment involved in serious incidents must not be tampered with and if necessary will be quarantined by Medical Physics. Incidents requiring external reporting to HFS or the MHRA will be managed in line with the Hazard and Safety Action Notice Policy. All external reporting must be channelled via the Equipment Coordinator (Head of Medical Physics).
CS-MPH-POL-5, Hazard and Safety Action Notice Policy
Evaluations
Equipment evaluation is a standard part of selection and procurement, and is encompassed by the Board’s policy.
CS-MPH-POL-10, Medical Equipment Funding and Acquisition Policy
The policy ensures equipment is safe, indemnified and meets regulatory standards (is CE Marked).
Clinical research
The process for controlling equipment required for clinical research is managed by the Research and Development Department
Non CE marked devices
All medical devices placed on the market in the UK and European Union must bear the CE Marking to demonstrate they meet the essential requirements of the Medical Device Directive. This helps ensure that they do not compromise the safety and health of patients, users and other persons when properly installed, maintained and used in accordance with their intended purpose.
The use of non CE marked devices is strictly prohibited within the NWTC Board unless carried out under a formal research programme, managed by Research and Development. It is a legal requirement that the UK competent authority (the MHRA) is informed of any clinical investigation involving a non-CE-marked medical device within the UK. These requirements are managed by the Research and Development Department and include:
- MHRA Notification and confirmation letter of no objection
- Clinical Investigation Plan
- Research Ethics Committee approval
- Adequate training for equipment users
- Systems for control and segregation of non CE marked equipment limiting usage to named clinical investigators
- Signed informed patient consent
- Criteria for stopping or curtailing use
Under all other circumstances, non CE marked medical equipment must not be used.
Off label usage of medical equipment and user modifications
Use of a medical device for anything other than its intended purpose is referred to as ‘off label’.
- Medical Devices within the NWTC Board must NOT be used off label.
- Medical Devices within the NWTC Board must NOT be modified by users
The Medical Devices Regulations stipulate that the manufacturer of a device is responsible for establishing that the device is safe and that it is suitable for its intended purpose. To establish this, manufacturers implement appropriate controls on the device design and manufacture and evaluate the safety and performance of the device in its intended application. This involves an analysis of risks that could arise during use, an assessment of relevant pre-clinical and clinical data, the preparation of appropriate instructions for use and, if necessary, specific training schemes. From such activities, manufacturers are able to verify that risks have been eliminated or minimised and are judged acceptable when weighed against the anticipated benefits to patients.
Specific guidance on the off label use and user modification of CE marked medical devices is provided by the MHRA in MDA/2010/001.
http://www.mhra.gov.uk/home/groups/dts-bs/documents/medicaldevicealert/con068160.pdf
Important points include:
- The use of a device in these circumstances exposes users and patients to unknown and therefore unacceptable risks and may have legal and ethical implications
- As well as the risks to the patient and user, liability for the performance and safety of products that have been modified, adapted or used off-label, could be transferred to the user
- Ensure that you are familiar with the instructions for use including the intended purposes for all the devices you use
- Only use devices for their intended purpose; do not modify or alter the function or structure of medical devices unless specifically sanctioned by the instructions for use
- Where a healthcare professional judges there is no alternative to off-label device use, the patient must be fully informed during the consent procedure and a note made in the patient's records
Exceptional circumstances
Under exceptional circumstances it is conceivable that users may be presented with a situation where no commercially available medical devices are available that are CE marked for the required application. Under these circumstances a formally documented detailed risk assessment will be required. This will include input from Medical Physics, Materials Management, the equipment manufacturer and have signoff by the Medical Director. Documented patient consent will be required along with a note in the patient’s record.
Prescribing medical equipment
Any professional user who prescribes medical equipment for use by a patient must be qualified to do so. Non-qualified staff who issue equipment must have the necessary written authority from a professional user before releasing the equipment or have been deemed competent to release it themselves.
Managers must ensure that medical equipment is not issued to patients or carers without the issue of the appropriate instructions and training and having ensured that facilities for maintenance and repair have been clarified.
Accreditation
All medical equipment lifecycle management functions carried out by the Medical Physics department are performed to the ISO 13485 Quality Management Standard. This is certified by an independent body on behalf of UKAS, the sole national accreditation body recognised by government to assess against internationally agreed standards.
ISO 13485, Medical devices – Quality management systems – Requirements for regulatory purposes, is an internationally agreed standard that sets out the requirements for a quality management system specific to the medical devices industry. The standard represents an international consensus on good quality management practices. The application of the ISO 13485 standard in itself ensures that all internal processes minimise risk.
Compliance with a formal quality management system is a recommendation of the MHRA in Managing Medical Devices 2021 and is required in NHS Scotland as detailed in both CEL35 and SHTN-00-04.
Audit and monitoring
Monitoring the organisation’s medical device management performance is essential to minimise or eliminate risks to patients and staff. Patient safety is enhanced by the use of systemic activities that prevent or reduce the risk of harm to patients. The Board assesses the efficacy of these arrangements through the following mechanisms:
- Annual Internal audit of all equipment management processes
- Annual independent audit of the quality management system to the ISO 13485 standard
- Feedback from other auditing activities (example: NHS QIS, Anaesthesia – Care Before, During and after Anaesthesia)
- Review of medical device incidents
Outsourcing
All aspects of the medical equipment lifecycle management process are managed by the Board. No processes are directly outsourced. Service contracts exist for some medical equipment and co-operative agreements with suppliers are sometimes utilised. In all instances management of the equipment is coordinated directly by Medical Physics.
